

pibase tools for validational and comparative analysis of BAM files
pibase is an open-source package of linux command line tools for validating next-generation sequencing loci (SNPs and loci of interest where no SNPs are known) and for comparative analyses using Fisher's exact test.
Acknowledgement: The development of pibase was partly funded by: The German Ministry of Education and Research (BMBF); the National Genome Research Network (NGFN); the Deutsche Forschungsgemeinschaft (DFG) Cluster of Excellence 'Inflammation at Interfaces'; the EU Seventh Framework Programme [FP7/2007-2013, grant numbers 201418, READNA and 262055, ESGI].
Disclaimer: pibase is provided free of charge for non-commercial use but you are required to read our disclaimer and to cite us when publishing results.
Download: pibase 1.4.7 example data (12GB) example output only (130kb)
Overview
| pibase |
Acronym for: get Position Information at BASE position of interest.
|
| Interoperability |
Input and output file types.
|
| Work flows |
Preparing BAM-files and using the pibase tools.
|
| QuickStart Tutorial | Prerequisites, installation, and pibase examples using BAM-files from the 1000 Genomes project. |
|
Essentials: |
|
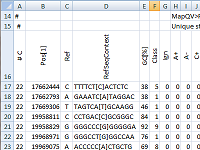
| pibase_bamref | Extract information from a BAM-file and a reference sequence file and table this information into a tab-separated text file. |
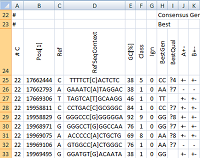
| pibase_consensus | Infer 'best' genotypes and their 'quality' classification, and optionally merge multiple pibase_bamref files (e.g. a control panel or several runs of the same patient) into a single file. |
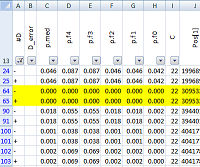
| pibase_fisherdiff | Compare two pibase_consensus files using Fisher's exact test on original data (aligned reads) rather than comparing processed data (SNP-calls or genotypes). |
|
Annotate: |
|
| pibase_tosnpacts | Works with our unpublished annotation pipeline. Contact us if you wish us to annotate your pibase files with our annotations. |
| pibase_annot | Works with our unpublished annotation pipeline. Contact us if you wish us to annotate your pibase files with our annotations. |
| pibase_tag |
Add primer region tags to a pibase_bamref, pibase_consensus, pibase_fisherdiff, or pibase_annot file.
|
|
Phylogenetics: |
|
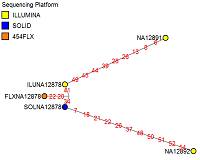
| pibase_to_rdf | Generate rdf file (for phylogenetic network analysis) from set of pibase_fisherdiff files. |
| pibase_rdf_ref | Generate a reference sample file from a pibase_consensus file, prior to generating the set of pibase_fisherdiff files required for pibase_to_rdf. |
| pibase_chrm_to_crs | Extract Cambridge Reference Sequence (Anderson 1981) variants from reads mapped to chrM (hg18, hg19, or NCBI36). |
|
Utilities: |
|
| pibase_to_vcf |
Convert a single-sample pibase file into VCFv4.1 format.
|
| pibase_c_to_contig |
Convert pibase-contig-numbers into contig names (e.g. 25 -> chrM).
|
| pibase_flagsnp |
Flag non-reference genotypes in a pibase_consensus file as potential mismatches ("SNPs" in NGS-parlance).
|
| pibase_diff |
Compare two pibase_consensus files using (BestGen) genotypes and a (BestQual) quality threshold.
|
| pibase_gen_from_snpacts | Works with our unpublished annotation pipeline. Contact us if you wish us to merge SNPs from SNP-callers, SNP-chips, or Sanger sequences into your pibase files for a genotype-comparison. |
| pibase_ref | Gets 500 flanking nucleotides of reference sequence around each coordinate (of the input list) and outputs a pseudo-FASTA file. For example for manual Sanger-sequencing primer design. |
Interoperability
pibase reads genomic coordinates of interest from a VCF*, samtools pileup, SOLiD Bioscope gff3, or a tab-separated file.
pibase extracts data at the coordinates of interest from an indexed FASTA reference and from a BAM-file** generated by BFAST, BWA, SSAHA2, samtools, SOAP (after conversion using soap2sam.pl), and SOLiD Bioscope. To extract the most complete information (including homologous region information and low-coverage genotypes), please use the raw unfiltered BAM-file (which includes non-uniquely mapped reads and duplicate reads).
pibase outputs tab-separated text files which can then be used in popular spreadsheet software, or filtered from the linux command line using grep, awk, and cut. pibase can also output variants into VCF, rdf, and snpActs formats.
*VCF is the variant list format accepted by European Nucleotide Archive. (VCF files are e.g. generated by GATK and samtools.)
**BAM is the sequence format preferred by the European Nucleotide Archive.
Work flows
"Essentials" work flow
-
Prepare an indexed sorted BAM-file with MD tags using samtools: Create BAM file with MD tag.
If you have several BAM-Files for the same sample, and some BAM-files were generated from the same library: merge all BAM-files from the same library into one BAM-file (because pibase_consensus counts the number of unique start points independently per BAM-file).
[samtools sort -o unsorted.bam > bamfile.bam]
samtools calmd -b bamfile.bam referencefile.fasta > bamfile.md.bam
[samtools merge outfile.bam infile1.bam infile2.bam [...]]
samtools index bamfile.md.bam - pibase_bamref : extract position info from BAM file and reference sequence file.
- pibase_consensus over single run: infer multi-filter-level genotypes from a single pibase_bamref-file and classify the genotypes into stable or dubious genotypes (BestQual flag).
- pibase_consensus over multiple runs: infer multi-filter-level ''consensus'' genotypes from pibase_bamref-files from multiple runs and classify the genotypes into stable or dubious genotypes (BestQual flag).
- [Optional: pibase_fisherdiff : compare two samples by unique start point counts (Fisher's exact test 2x4), using the pibase_consensus-files]
- First, carry out the "Essentials" workflow, in order to get a pibase_consensus file or a pibase_fisherdiff file.
- [Optional: pibase_tosnpacts : export genotypes from pibase_consensus or pibase_fisherdiff or pibase_diff to snpact-format (snpact -ft 'own' -sp 1).
- [Optional: pibase_annot : annotate pibase_diff files with rsID, gene name, and other information]
- [Optional: pibase_tag : for PCR enriched samples, tag snps which are in PCR primer regions +- 1 base, using pibase_consensus-files or pibase_diff-files or pibase_annot files]
- First, carry out the "Essentials" workflow, in order to get a set of pibase_consensus files.
- [Optional: pibase_rdf_ref : create a reference sample from one of the pibase_consensus files.]
- Create a tab-separated text file detailing the sample files in the group, and the sample names which should be displayed in the phylogenetic network (see pibase_to_rdf). The first sample in this text file can either be one of the group of samples, or the reference sample created by pibase_rdf_ref.
- pibase_to_rdf : create an rdf file from a set of pibase_fisherdiff files for subsequent phylogenetic network analysis, e.g. evolutionary analysis, or sample mix-up (or confusion) analysis.
- [Optional: pibase_chrm_to_crs : extract Cambridge Reference Sequence (Anderson et al., 1981) variants from reads mapped to chrM (hg18, hg19, or NCBI36)]
- First, carry out the "Essentials" workflow, in order to get a pibase_consensus file or optionally a pibase_fisherdiff file.
- [Optional: pibase_to_vcf : From a pibase_consensus file, create a VCF file that can e.g. be submitted to the European Nucleotide Archive or processed with VCFtools.]
- [Optional: pibase_c_to_contig : Convert pibase-contig-numbering (e.g. in a pibase_consensus file or a pibase_fisherdiff file) to contig names, e.g. 25 to chrM.
- [Optional: pibase_flagsnp : Flag non-reference genotypes in a pibase_consensus file as potential mismatches ("SNPs" in NGS-parlance), using the pibase_consensus-files, pibase_fisherdiff-files or annotated versions of these files.]
- [Optional: pibase_diff : compare two samples (conventionally) by genotypes, using the pibase_consensus-files. This is much less accurate than pibase_fisherdiff and only included for those users interested in a conventional comparison!]
- [Optional: pibase_gen_from_snpacts : Merge SNPs from SNP-callers, SNP-chips, or Sanger sequences into pibase_consensus, pibase_fisherdiff, or annotated versions of these files. The idea is to generate a side-by-side genotype and nucleotide signals comparison table.]
QuickStart Tutorial
Prerequisites
- Linux operating system (we use CentOS 5.5 / linux 2.6.18-194.32.1.el5 on our linux cluster and Ubuntu 8, 9, or 10 on our PCs.)
- Python v2.4.3 or v2.6.5 or v2.7.2 (recommended) (usually already installed on linux clusters or linux PCs)
- pysam v0.6 (if using other versions, test for pysam bugs using large data sets)
- GNU Fortran (usually already installed on linux clusters, or installable using the Synaptics package manager under Ubuntu PCs)
- 1GB of RAM (2GB for pibase_fisherdiff)
- Bash command line, or a linux cluster job scheduler such as PBS.
Installation
Download pibase.v1.4.7.tar.gz and unzip.
From the linux command line, navigate into the pibase directory and enter ./f.sh to compile the pibase_643 subprogram.
Finally, include the pibase directory in your linux PATH variable.
Test run
Test whether the installations were successful using our example data (12GB size!): Download the 12GB tar.gz file and unzip. From the linux command line, navigate into the example data folder and then run the example shell script by entering
./pibase_test.sh
(If problems occur, check the pre-requisites).
When pibase_test.sh runs successfully, it creates a directory called output, into which the results files are written.
Next, run the phylogenetic example shell script by entering
./pibase_to_rdf.sh
To validate the test runs, compare directories output and output_validated from the linux command line:
diff output output_validated
Windows users
Windows users (and impatient Linux users) can download just the small zipped output_validated folder (130kb) for a quick impression of the output files. Import these tab-separated text files into Excel for viewing, filtering, or sorting. Note that there are floating point and floating comma versions of the files, the latter of which are generated using simple linux commands in the example shell script pibase_test.sh.
Detailed examples
Please look into the shell script pibase_test.sh and pibase_to_rdf.sh web pages which give several examples how to start the pibase tools.
Essentials | pibase_bamref
pibase_bamref extracts information at genomic coordinates of interest from a BAM file and a reference sequence file and tables this information into a tab-spearated text file. Optionally, information can be displayed on the command line. A single genomic coordinate can be specified on the command line, or a list of coordinates can be specified in a file. Note that BAM-files must include MD-tags.
Usage:
pibase_bamref [-[v][d][p chr]] {table or poi} ref bam out LR
QVmin [[[MLmin] [[MMmax] [RQVmin] [maxr]]]]
[] optional
-vdp [v]erbose output into terminal,
[d]etailed list of reads,
at single [p]osition: chr poi
chr: chromosome name (or contig name)
poi: position of interest (chromosomal coordinate, 1-based)
table: file of poi's: vcf, gff3, pileup,
or plain tab-file: chr TAB poi LINEBREAK
The format is detected by pibase_bamref. If the plain tab-file is used,
the lines must be sorted by chr-names, and the chr-names must be ordered
in the sequence of the reference sequence file or BAM-file-header.
ref: reference sequence file
bam: sorted bam file, bam index file must also exist
out: name for (tab-separated) output file
LR: max length of reads (e.g. 50)
QVmin: optional min base quality (default: 20)
MLmin: optional min mapped read length (default: 49)
MMmax: optional max number of base space mismatches in read (default: 1)
RQVmin: optional min read mapping quality (default: 20)
maxr: optional max number of reads considered at a poi (default: -1)
(default: unlimited. For ultra-high coverage files:
use an integer value of e.g. 1000 as the limit).
Examples:
pibase_bamref -vp chrM 16189 hg20.fasta na12752.bam na12752_chrM_16189.txt
pibase_bamref list.txt hg18.fasta na12752.bam na12752_list.txt
pibase_bamref pileupsnps.txt hg18.fasta na12752.bam na12752_pileupsnps.txt
pibase_bamref gatksnps.vcf hg18.fasta na12752.bam na12752_gatksnps.txt
Notes:
If you are generating your own input lists with positions of interest: The genomic coordinates must be 1-based (the UCSC browser, VCF SNPlists, samtools pileup SNPlists, and Bioscope SNPlists use 1-based coordinates). SNPlists from Bioscope 1.2.1 or samtools pileup can be used without checking - these are ok.
pibase_bamref speed depends on depth of coverage and read length, and on python/pysam version. For high coverage BAM-files (50x-100x) and 50bp read lengths, pibase_bamref v1.4.5 processes about 50,000-100,000 genotypes per hour on a single core using 2GB RAM . At least python v2.7 and at least pysam v0.5 are required for this speed, otherwise pibase_bamref is up to 100x slower. As there are critical but silent bugs in other pysam versions, please do not use other pysam versions unless you test that version thoroughly (e.g. by running our chr22 and exome example data sets and performing a diff)!
For the most complete information: give pibase the original BAM file (which should include the ambiguously mapped reads, and which can also include "duplicate" reads)
Essentials | pibase_consensus
pibase_consensus adds information to a pibase_bamref file, inferring 'best' genotypes and 'best qualities', and optionally merging read counts from multiple pibase_bamref files (e.g. a control panel or several runs of the same patient) into a single pibase_consensus file. A BestQual question mark ("?") indicates that the BestGen is instable with respect to filtering, and therefore should not be used without further validations.
Usage:
pibase_consensus [-t[h][v [ver]] t1 t2 t3 t4 t5 {h1 h2}] in1 [in2 ... inN] out
[]=optional {}=only for "-th" option
[-t[h][v] (optional) minimal thresholds for allele calling (A, C, G, or T),
(optionally) followed by "h" to override h1 and h2 defaults,
(optionally) followed by "v" to revert to an older version
[ver] (optional) older pibase_consensus version to revert to.
To list all available older versions, specify "-tv" without ver
(older version 1.4.3: arguments h1 and h2 not available)
t1 min fraction of reads indicating allele (default: 2.2%, i.e. 0.022)
t2 min number of reads indicating allele (default: 8)
t3 min fraction of unique start points indicating allele (default: 0.04)
t4 min number of unique start points indicating allele (default: 4)
t5 both-stranded confirmation only if, for at least one filter level,
min-strand-counts >= t5 * max-strand-counts (default t5: 0.2)
{h1 min fraction of unique start points that need to be different between
filter F2-F3 resp. F3-F4 in order to flag a hypervariable resp.
homologous locus (default: 2.2%, i.e. 0.022)
h2} ] min number of unique start points difference between F2-F3 resp. F3-F4
in order to flag a hypervariable resp. homologous locus (default: 2)
inX input file[s] = pibase_bamref output file[s]
must have same reference sequence (not checked!)
and same positions of interest
out name for (tab-separated) output file
Examples:
# Single file
pibase_consensus na12752_pileupsnps.txt genotypes_na12752_pileupsnps.txt
# Two files
pibase_consensus na12752_1.txt na12752_2.txt gen_na12752_1to2.txt
# Decreased sensitivity to sequencing or mapping errors or contamination
pibase_consensus -t 0.1 8 0.15 8 0.01 na12752_pileupsnps.txt genotypes_na12752_pileupsnps.txt
Notes:
What pibase_consensus does:
- Reads the pibase_bamref file and adds further columns of information: For each genomic coordinate of interest: 10 rule-based genotype decisions and one "BestGen" (best genotype) from these 10 genotypes, strand support for the A and B alleles of the "BestGen", and summary allele counts for each of the 10 genotypes.
- If multiple pibase_bamref files are specified, consensus genotypes are computed for pooled allele counts over all files (e.g. for a library which was sequenced in multiple runs or multiple lanes). The output file format is identical to the single-file output except that all specified input files are listed the file header.
- Low quality genotypes are annotated with BestQual tags ?1 to ?8; where ?1 denotes not too bad quality and genotypes with higher values should be rejected or inspected further. If there is no tag, the genotype is assumed to be validated by all 10 filter methods at a minimal coverage of t2 (default: 8) per allele of which there are at least t4 (default: 4) unique starting points per allele. The quality grading aims to help you understand the underlying mechanisms of each problem.
- Both-strandedness confirmation must be applied using the additional tags A+- and B+-. (Because some enrichment methods are strand-biased and occasional failure of both-stranded support is common in next-generation sequencing, we did not bundle the strandedness criterion into the BestQual tag).
- For slightly higher specificity, the SNVs near indels can be filtered using the additional tag Ign.
- Regions of simple repeats can be filtered using the Class tag (>1), regions of segmental duplications can be filtered using the Homologous tag ("H"), and hypervariable regions can be filtered using the Hypervariable tag ("V").
| ?1 : | Slightly poor genotype quality. Mapping stringency versus reference sequence context class looks ok. Not all 10 genotyping filter stages lead to the same genotype. However, for the high mapping stringency filter stages, at least t4 (default: 4) unique start points and at least t2 (default: 8) reads support this genotype. |
| ?2 : | Somewhat poor genotype quality. Mapping stringency versus reference sequence context class looks ok. This genotype is supported by less than 5 filter stages, but by at least 2 filter stages, of which one stage is in the unique start points category, and the other stage is in the coverage category. |
| ?3: | Really poor quality. Reference sequence context looks difficult (homopolymeric run>4, or STRs) and mapping stringency was low. But at least one stringent filter supports this genotype. |
| ?4: | Very poor quality. Reference sequence context looks difficult (homopolymeric run>4, or STRs) and mapping stringency was low. But at least one of the unique-start-point filters supports this genotype. |
| ?5: | Highly problematic quality. The best unique-start-point derived genotype is in conflict to the best coverage-derived genotype. |
| ?6: | Highly problematic quality. The best unique-start-point derived genotype is in conflict to the best coverage-derived genotype, and the best coverage-derived genotype is "senior" to the best usp-derived genotype. |
| ?7: | Low-coverage guess. The coverage is below t2 (default: 8) (can be as low as 1). |
| ?8: | Low-coverage guess. The coverage is below t2 (default: 8) (can be as low as 1). Reference sequence context looks difficult (homopolymeric run>4, or STRs), and there are no stringently mappable reads. |
Poor quality genotypes and single-strand genotypes can be filtered using linux commands as follows:
# copy header lines into new file:
grep '^#' pibase_consensus_file > validated_pibase_consensus_file
# append filtered table lines (indel-reads < 3, tags A+- and B+- and BestQual) to the new file:
awk 'BEGIN {FS="\t"};($7<3)&&($10=="+-")&&($11=="+-")' pibase_consensus_file | grep -v '?' >> validated_pibase_consensus_file
Potentially problematic regions can be filtered or counted using linux commands as follows:
# count number of genotypes in homologous loci:
grep -v '^#' pibase_consensus_file | grep 'H' | wc -l
# count number of genotypes in hypervariable loci:
grep -v '^#' pibase_consensus_file | grep 'V' | wc -l
# count number of genotypes near indels:
awk 'BEGIN {FS="\t"};($7>0)' pibase_consensus_file | wc -l
Essentials | pibase_fisherdiff
pibase_fisherdiff merges two pibase_consensus files and tags differences between these two files (e.g. control sample and case sample) based on Fisher's exact test for five different filter levels. (See example.)
Usage:
pibase_fisherdiff control case out [[cov] [[p] [[fac]]]
control: pibase_consensus file of control (healthy) sample
case: pibase_consensus file of case (diseased) sample
out: output file
[] optional
[cov]: min coverage threshold (optional. Default: 100)
[p]: max median p-value threshold (optional. Default: 0.01)
[fac]: max factor (optional. Default: 3.5).
Example:
# min coverage 50, p <= 0.01, fac <= 10
pibase_fisherdiff normal.txt tumor.txt diff_out.txt 50 0.01 10
Notes:
Eight leading columns are added to the pibase_consensus output, and the two samples are merged into one file which can then be filtered using grep or awk or python/perl/java/etc scripts, or imported into Excel by Windows users and viewed/filtered further in Excel:
-
In the leading column, significant similarity at a genomic coordinate is denoted by "=-" (control sample) or "=+" (case sample).
Significant difference at a genomic coordinate is denoted by "-" (control) and "+" (case).
Low coverage in one or both samples is denoted by "?-" and "?+". - For those who want to understand the underlying mechanism: The control and case samples are computed to be significantly different at a genomic coordinate if the median p-value <= p and the coverage in each sample >= cov and the factor <= fac. The factor is defined as follows: If the major alleles are identical in case and control, major allele counts / minor allele counts must be <= fac; if the major alleles are different, the factor criterion is not used. The coverage and factor criteria must be applied because the p-value changes quite a lot at coverages of about 50 or lower, when just 1 read is shifted from the major allele to the minor allele (i.e. when a normal sequencing error occurs). In other words, for special cases the p-value alone is not a sufficiently accurate indicator of similarity or difference.
-
In the second column, the tag D_error indicates whether the (default or user-specified) thresholds for the applicability of Fisher's exact test have been violated.
In detail, if the coverage falls below the threshold, D_error at the genomic coordinates displays an '!' and bit 0 (control) or 1 (case) is set. If the major/minor allele counts threshold is exceeded, D_error is output with an '!' and bit 2 (control) or 3 (case) is set. For example '!1' indicates low coverage in the control sample, and '!12' indicates the major/minor allele threshold violation in both samples. - Fisher's exact test p-values (two-tailed) are given in column 3 (median p-value) and columns 5-8 (for each of the 5 filter levels, computed for ACGT counts of unique start points).
- Both-strand-support testing is not included in the first 3 columns, so explicit filtering using the A+- and B+- tags should be performed where considered, see pibase_consensus filtering.
- Mathematical detail: The p-values are computed with ACM algorithm 643 (http://portal.acm.org/citation.cfm?id=214326) for a 2x4 matrix using a hybrid approximation. This network method overcomes numeric overflow problems associated with online web calculators.
- Compute speed: about 20,000-50,000 genomic coordinates per hour on a single core using 2GB RAM.
# copy header lines (i.e. starting with '#') into new file:
grep '^#' pibase_fisherdiff_file > filtered_file
# append filtered table lines to the new file (p-value <= 0.005, and tags A+- and B-):
awk 'BEGIN {FS="\t"};($3<=0.005)&&($18=="+-")&&($19=="+-")' pibase_fisherdiff_file >> filtered_file
# count differences:
grep '^+' filtered_file | wc -l
Annotate | pibase_tosnpacts
pibase_tosnpacts reads the "BestGen" genotypes from a pibase_consensus file or a pibase_fisherdiff file and creates a file (or file pair, if pibase_fisherdiff) for import into snpacts. After annotation within snpacts, the snpacts-annotations are exported and added to the pibase_consensus or pibase_fisherdiff file using pibase_annot.
Usage:
pibase_tospnacts in out1 [out2]
in: file from pibase_consensus or pibase_fisherdiff
out1: output file (for input into snpacts, in 'own' format)
out2: 2nd output file (case sample) if pibase_fisherdiff
Examples:
# pibase_consensus file:
pibase_tosnpacts geno_na12752.txt for_snpacts_na12752.txt
# pibase_fisherdiff file:
pibase_tosnpacts diff.txt for_snpacts_control.txt for_snpsacts_case.txt
Notes:
How to import the pibase_tosnpacts file into snpacts:
snpact -ft own -sp 1 -pchr 0 -psnp 1 -prb 2 -pbb 3 -psb 4 -pmt 5 -all -hg hg18 -t tablename filename
Remove problematic EOLs from BLOBs in annotated table:tableupdate -eol tablename
Export the annotated, de-BLOBBed table to csv:outputact -t tablename -based 1 -csv -worst -hg hg18 inputfilename outputfilename
where inputfilename is the file from pibase_tosnpacts, and outputfilename is the file for pibase_annotAnnotate | pibase_annot
pibase_annot annotates a pibase_consensus or pibase_fisherdiff file with snp information from an exported snpacts table (see above section on pibase_tosnpacts).
Usage:
pibase_annot type an1 [an2] in out
type: 1=pibase_consensus; 2=pibase_fisherdiff
an1: snpacts-annotations file 1 (control sample, created by outputact)
an2: snpacts-annotations file 2 (case sample, created by outputact)
annot2 is not specified for pibase_consensus (type=1)
in: tab-separated file from pibase_consensus or pibase_fisherdiff
out: results file with merging of annotations and infile
Examples:
# pibase_consensus file:
pibase_annot 1 annot.txt geno_na12752.txt anno_geno_na12752.txt
# pibase_fisherdiff file:
pibase_annot 2 anno_control.txt anno_tumor.txt diff.txt anno_diff.txt
Annotate | pibase_tag
pibase_tag adds primer region tags to a pibase_consensus, pibase_fisherdiff or pibase_annot file. The pibase file is annotated using information from a primer region file.
Usage:
pibase_tag typ pr in chr pos out [tag]
typ: type of input file (always enter 0)
pr: primer region file, tab-separated: chr pos start end strand (int, 1-based)
in: tab-separated file, e.g. from pibase_diff or pibase_annot
chr: column number (1-based) in in-file with chromosome number
pos: column number (1-based) in in-file with position
out: output file ( = tagged input file)
tag: (optional, default=column 2) column number, at which the 4 primer tag columns are inserted.
Example:
# pibase_annot file:
pibase_tag 0 primer_regions.txt pibase_annot.txt 4 5 pibase_tag_annot.txt 2
Notes:
The primer region file must have a special table header (see ## in example below) and is tab-separated:
chromosome, start, end, strand (integer, 1-based).
#dummy test file for pibase_tag ##chr start end strand 1 152644520 152644530 1 1 152644740 152644750 -1
Phylogenetics | pibase_to_rdf
pibase_to_rdf generates an rdf file (for phylogenetic network analysis) from a set of pibase_fisherdiff files.
Usage:
pibase_to_rdf list rdf [[pmax] [both] [elim]]
list: text file which lists the set of pibase_fisherdiff files:
file_name TAB sample_name EOL
rdf: name of output file (for analysis using Network)
[]: optional arguments:
[pmax]: max p-value to consider (default=0.01)
[both]: bothstranded confirmation (y/n, default=y)
[elim]: eliminate "N"-columns and invariable columns (y/n, default=n)
Example: See pibase_to_rdf.sh
Note:
The example above refers to our pibase paper. Please consult this paper if you are unfamiliar with phylogenetic network analyses.
Phylogenetics | pibase_rdf_ref
pibase_rdf_ref generates a reference-base pibase_consensus file which may be used as the reference sample for pibase_fisherdiff comparisons in a group of samples, e.g. for a pibase_to_rdf run and subsequent phylogenetic network analysis.
Usage:
pibase_rdf_ref in out [cov]
in: pibase_consensus file (any sample from the group of samples)
out: pibase_consensus file with genotypes taken from Ref column of in-file
cov: coverage (optional. default=50. Divisible by two.)
Example: See pibase_to_rdf.sh
Note:
The example above refers to our pibase paper. Please consult this paper if you are unfamiliar with phylogenetic network analyses.
Phylogenetics | pibase_chrm_to_crs
pibase_chrm_to_crs extracts Cambridge Reference Sequence (CRS)* variants from chrM** alignments. The CRS numbering is identical to the numbering of the revised CRS (rCRS)***. The reference sequence bases within the mtDNA control region are identical for the CRS and the rCRS.
(In hg18, hg19, and NCBI36, chrM is used as the mitochondrial reference genome; in NCBI build 37 rCRS is used and in hg20, rCRS will be used; most mtDNA databases are based on the CRS/rCRS genomic coordinates.)
Usage:
pibase_chrm_to_crs in out own pro sam [[t] [min]]
in: pibase_consensus genotype file (MUST be chrM ONLY!)
out: base name of output files. The following files are output:
out_f4maj.txt: major variants for highest-stringency filter (f4)
out_f4min.txt: minor variants for highest-stringency filter (f4)
out_f0maj.txt: major variants for lowest-stringency filter (f0)
out_f0maj.txt: minor variants for lowest-stringency filter (f0)
out_all.txt: complete pibase_consensus file in CRS notation
out_sum.txt: summary file in CRS notation
own: abbreviation of owner/PrincipalInvestigator of the project
pro: for summary: project abbreviation
sam: for summary: sample abbreviation
[]: optional arguments:
[t]: threshold for minor allele cut-off
(default: 0.015, i.e. 1.5% of total read counts at a given coordinate)
[min]: min number of read counts required for an allele-call (default: 2)
Example:
pibase_chrm_to_crs pat123_chrM.txt pat123_chrs.txt FRA fro Pat123
Note:
The pibase_consensus file must contain only chrM lines. If there are other lines in the file, the header lines and the chrM lines must be extracted before using pibase_chrm_to_crs:
# copy header lines into new file:
grep '^#' pibase_consensus_file > chrM_pibase_consensus_file
# append lines with "chromosome 25" (i.e. chrM) to the new file:
grep '^25' pibase_consensus_file >> chrM_pibase_consensus_file
References:
* Anderson, S., Bankier, A.T., Barrell, B.G., de Bruijn, M.H., Coulson, A.R., Drouin, J., Eperon, I.C., Nierlich, D.P., Roe, B.A., Sanger, F., Schreier, P.H., Smith, A.J., Staden, R., Young, I.G. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457-465.
** Ingman, M., Kaessmann, H., Pääbo, S., Gyllensten, U. (2000) Mitochondrial genome variation and the origin of modern humans. Nature, 7, 708-713.
*** Andrews, R.M., Kubacka, I., Chinnery, P.F., Lightowlers, R.N., Turnbull, D.M., Howell, N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet., 23, 147.
Utilities | pibase_to_vcf
pibase_to_vcf converts a single-sample pibase file into VCFv4.1 format.
Usage:
pibase_to_vcf [-h] [-v] [-d] [-u s s s s s s s s s s] in out nam bam
in: tab-separated file (pibase_consensus)
out: VCF file
nam: name of sample in VCF file (column header)
bam: name of bam file from which in file was generated
Example:
pibase_c_to_contig in.txt out.txt mysample mysample.bam
Utilities | pibase_c_to_contig
pibase_c_to_contig converts pibase-contig-numbers into contig names (e.g. 25 -> chrM).
Usage:
pibase_c_to_contig in [in2] out
in: file to be converted (from pibase_consensus, pibase_fisherdiff, ...)
Note: pibase_fisherdiff has a shortened file head, so a second input file
with the conversion table must be given.
in2: file with contig numbering/name table in header
(required, if the first input file is a pibase_fisherdiff file)
out: output file
Examples:
pibase_c_to_contig gen.txt gen_contigs.txt
pibase_c_to_contig diff.txt table.txt diff_contigs.txt
Utilities | pibase_flagsnp
pibase_flagsnp flags a BestGen genotype as being an "NGS SNP" (i.e. a mismatch) if it is different to the reference base. Three columns are inserted before the "BestGen" column: "IsSnp" (1/0 flag), "SnpReads" (non-reference-allele coverage at filter level 0), and "CovAll" (coverage at filter level 0, consisting of all reads indicating A, C, G, or T but not N)
To sort the pibase_consensus "BestGen" genotypes into Non-Reference (i.e. SNP) and Reference.
Usage:
pibase_flagsnp in out
in: tab-separated file (pibase_consensus, pibase_fisherdiff, or pibase_diff)
out: results file with merging of annotations and infile
Example:
pibase_flagsnp gen_id100.txt snp_gen_id100.txt
Note: Outside the NGS world, "SNPs" include reference genotypes which are polymorphisms occurring in 1% or more of a population - this is often a matter of confusion between NGS and non-NGS specialists.
Utilities | pibase_diff
pibase_diff compares two pibase_consensus files using (BestGen) genotypes and a (BestQual) quality threshold. (Accuracy can be much lower than pibase_fisherdiff, i.e. up 10x or more false positives and false negatives, but compute time is fast.)
Usage:
pibase_diff control case out worst
control: pibase_consensus file of control (healthy) sample
case: pibase_consensus file of case (diseased) sample
out: output file
worst: worst BestQual level to consider (0-8)
Examples:
# Consider BestGens up to a worst BestQual of ?1 (i.e. ignore ?2 to ?8):
pibase_diff normal.txt tumor.txt diff_out.txt 1
Notes:
One leading column is added to the pibase_consensus output, and the two samples are merged into one file which can then be filtered using grep or python/perl/java/etc scripts, or imported into Excel by Windows users and viewed/filtered further in Excel:
- In the leading column, identical BestGen genotypes are denoted by "=-" (control sample) or "=+" (case sample).
- Different BestGen genotypes at a genomic coordinate are denoted by "-" (control) and "+" (case).
# copy header lines into new file:
grep '^#' pibase_diff_file > filtered_diff_file
# append filtered lines to the new file (filtering by first column and tags A+- and B+-):
awk 'BEGIN {FS="\t"};($11=="+-")&&($12=="+-")' pibase_diff_file | grep -v '^=' >> filtered_diff_file
# count differences:
grep '^+' filtered_diff_file | wc -l
Utilities | pibase_gen_from_snpacts
pibase_gen_from_snpacts inserts snpacts genotypes into a pibase file before the "BestGen" column. Multiple runs of this script can be performed to insert genotypes from several different sources (e.g. samtools, Bioscope, GATK). Only chromosomes chr1-chr22, chrX, chrY are recognised in snpacts files.
To compare genotypes from different sources with signals and genotypes from pibase_consensus.
Usage:
pibase_gen_from_snpacts typ s1 [s2] in out hdr
typ: type of input file (1=pibase_consensus or pibase_gen_from_snpacts)
s1: snpacts file1, created by outputact
s2: snpacts file2, (not specified for typ=1)
in: tab-separated file, from pibase_consensus or pibase_gen_from_snpacts
out: results file with merging of annotations and infile
hdr: header of inserted snpacts-column (e.g. samtools, Bioscope, GATK)
Example:
# add samtools SNPs into pibase_consensus file:
pibase_gen_from_snpacts 1 id100_samtools.txt gen_id100.txt sam_gen_id100.txt samtools
# add Sanger SNPs into samtools-pibase_consensus file:
pibase_gen_from_snpacts 1 id100_sanger.txt sam_gen_id100.txt san_sam_gen_id100.txt sanger
Utilities | pibase_ref
pibase_ref gets 500 flanking nucleotides of reference sequence around each coordinate (of the input list) and outputs a pseudo-FASTA file.
Primary use: manual Sanger-sequencing primer design.
Usage:
pibase_ref in ref out
in: file with list of genomic coordinates (e.g. a VCF file):
chromosome_name TAB 1_based_coordinate_in_chromosome
ref: indexed reference sequence file (fai must exist)
out: FASTA-formatted output file
Example:
pibase_ref snplist.vcf hg19.fa flanked_snps.fasta
Citing pibase
Please cite this website www.ikmb.uni-kiel.de/pibase as well as our paper
Forster, M., Forster, P., Elsharawy, A., Hemmrich, G., Kreck, B., Wittig, M., Thomsen, I., Stade, B., Barann, M., Ellinghaus, D., Petersen, B.S., May, S., Melum, E., Schilhabel, M.B., Keller, A., Schreiber, S., Rosenstiel, P., Franke, A.
From next-generation sequencing alignments to accurate comparison and validation of single nucleotide variants: the pibase software.
Nucleic Acids Res., doi: 10.1093/nar/gks836
Disclaimer, terms, and conditions
The software, documentation and any help or support are provided "as-is" and without any warranty of any kind. Installation of the required python module pysam and its dependencies must be carried out by a competent linux user or administrator at their own risk (installation of modules or dependencies can render previously working software or even the operating system unusable).
By downloading or using the pibase tools, the Non-Commercial User accepts these unmodified terms and conditions.
The term "Software" in these terms and conditions refers to the pibase software components, including upgrades, additions, copies and documentation as supplied by the Institute of Clinical Molecular Biology.
The term "Non-Commercial User" in these terms and conditions refers to the person who uses the Software for non-commercial purposes. Commercial use is prohibited unless expressly permitted in writing by the Institute of Clinical Molecular Biology.
The term "person" includes the organisation or group if the Software is used within the activities of an organisation or group.
The Institute of Clinical Molecular Biology grants to the Non-Commercial User a non-exclusive license to use the Software free of charge. The Institute of Clinical Molecular Biology grants to the Non-Commercial User a non-exclusive license to use, store, publish and exploit free of charge and without any time limit any results which the Non-Commercial User has generated with the help of the Software, provided that the Non-Commercial User cites the Software and version, the web site www.ikmb.uni-kiel/pibase and the paper on pibase.
The Non-Commercial User acknowledges that the Software is a tool to be used by the Non-Commercial User in his or her work and that the Non-Commercial User shall remain solely responsible for the content and quality of any results produced by the Non-Commercial User from the use or with the aid of the Software. The Non-Commercial User undertakes to indemnify and hold the Institute of Clinical Molecular Biology harmless in the event of any claim against the Institute of Clinical Molecular Biology arising from any incorrect or misleading information that the Non-Commercial User has obtained with the aid or by the use of the Software.
The Institute of Clinical Molecular Biology reserves all intellectual property rights of the source code and prohibits the copying, re-writing, or re-use of the code or parts thereof except for the purposes expressly granted in this agreement. These terms and conditions shall be governed by and construed and interpreted in accordance with German Law and submitted to the sole jurisdiction of the court of Kiel (Germany).
Contact
Michael Forster or Prof. Dr. rer. nat. Andre Franke.
m [dott] forster [att] ikmb [dott] uni - kiel [dott] de
a [dott] franke [att] mucosa [dott] de
Institute of Clinical Molecular Biology
Christian-Albrechts-University of Kiel
Schittenhelmstrasse 12
D-24105 Kiel
Germany
^top